


Summary
The Clinical Trials Information System (CTIS)sits at the intersection of EU CTR 536/2014 and the GDPR, creating layered dataprotection obligations for sponsors, the European Medicines Agency (EMA), theEuropean Commission and Member States. The Joint Controllership Arrangement(JCA), updated with effect from 12 March 2026, formalises how those obligationsare allocated. This article explains the legal architecture, identifies eachparty’s responsibilities and provides practical guidance for sponsors seekingto maintain inspection ready CTIS data protection compliance.
The iliomad CTIS Compliance Framework
iliomad approaches CTIS data protection using a structured methodology built on three pillars:
• Map: identify all personal data flows within and outside CTIS, including data shared with national competent authorities and ethics committees.
• Allocate: confirm controllership status for each processing activity using the JCA as the primary reference instrument.
• Operationalise: embed GDPR obligations (transparency, data minimisation, security and data subject rights) into trial documentation, including the protocol, the Informed Consent Form (ICF) and the Data Management Plan.
This framework ensures that compliance is not retrofitted at the end of a study but is built into its architecture from first submission.
The Legal Framework: EU CTR 536/2014, the GDPR and the JCA
The Clinical Trials Information System was established under Regulation (EU) No 536/2014 on clinical trials on medicinal products for human use (EU CTR), which replaced Directive 2001/20/EC and became fully applicable on 31 January 2022. CTIS serves as the single entry point for the submission, assessment and supervision of clinical trials across the European Economic Area.
Because CTIS processes personal data, including data relating to investigators, authorised representatives and, in certain configurations, trial participants, it falls within the scope of Regulation (EU) 2016/679 (the General Data Protection Regulation, GDPR). Where EMA and the European Commission process personal data in their capacity as EU institutions, Regulation (EU) 2018/1725 applies in parallel.
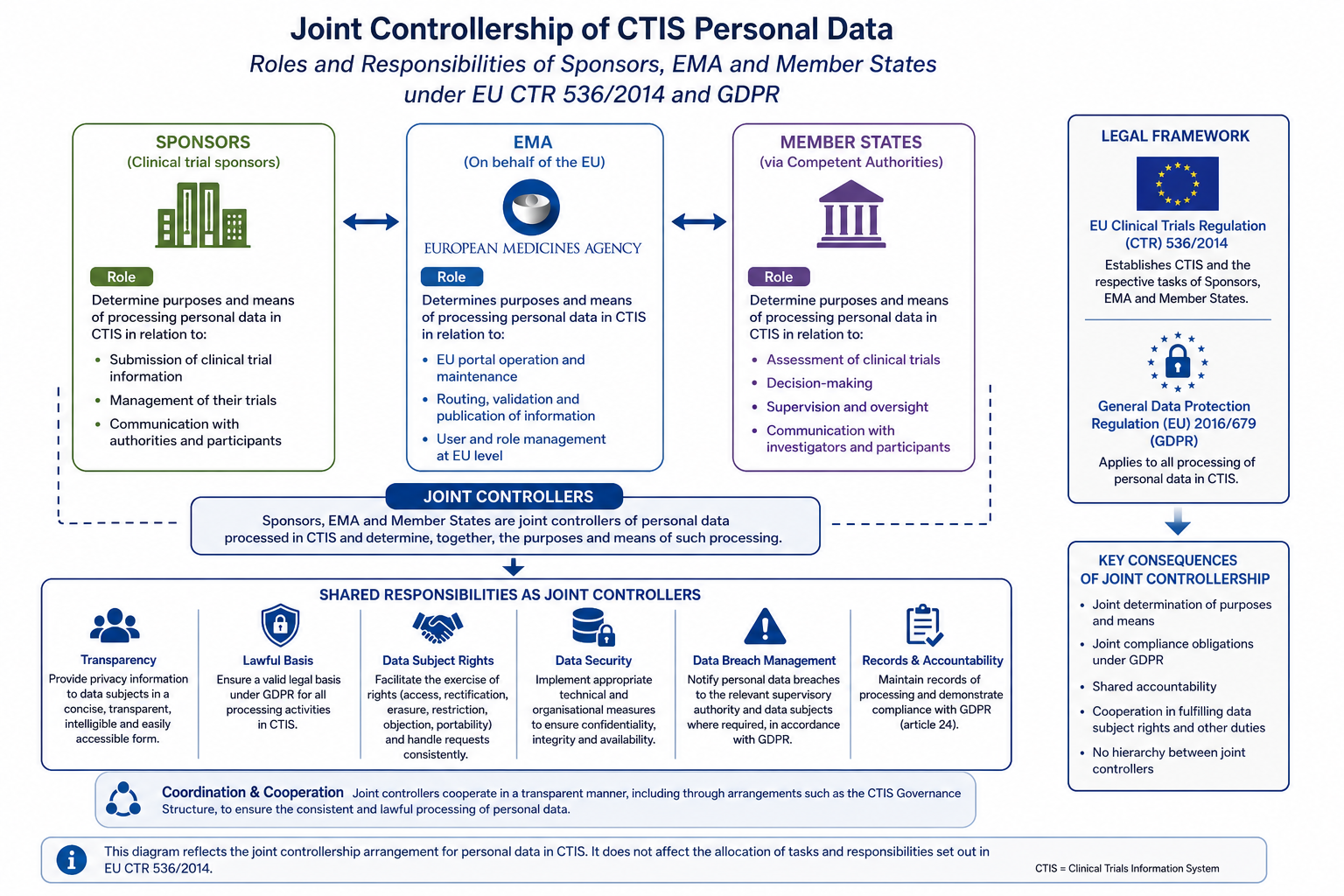
The Joint Controllership Arrangement is the instrument that operationalises Articles 26 and 28 of the GDPR within the CTIS environment. Updated with effect from 12 March 2026 following the implementation of the Revised CTIS transparency rules, the JCA clarifies which entity determines the purposes and means of each processing activity, thereby fixing liability and establishing the procedures that govern data subject requests and security incidents.
Focus: The European Medicines Agency (EMA)
EMA acts as a joint controller for those processing activities within CTIS that fall within its remit as system operator. Under the JCA, EMA is responsible for the technical and organisational security measures applied at platform level, for publishing transparency information on its website and for coordinating responses to data subject requests that originate from the central CTIS interface. EMA’s Data Protection Officer, appointed under Regulation (EU) 2018/1725, provides independent oversight of these activities and serves as the primary point of contact for the European Data Protection Supervisor (EDPS).
Focus: The European Commission
The European Commission retains joint controllership responsibilities relating to the policy framework that governs CTIS and certain data processing activities associated with the development and maintenance of the system’s technical infrastructure. The Commission is also accountable for ensuring that the JCA itself remains aligned with evolving GDPR guidance issued by the European Data Protection Board (EDPB).
Allocation of Roles Under the Joint Controllership Arrangement
The JCA distributes controllership among four principal categories of party: the European Commission, EMA, Member States (acting through their national competent authorities) and sponsors. Understanding which entity bears primary responsibility for a given processing activity is essential for drafting compliant trial documentation and responding to regulatory inspections.
The allocation of roles is as follows:
• European Commission: joint controller for infrastructure and policy (GDPR Art. 26 and Regulation 2018/1725).
• EMA: joint controller for platform operation and transparency (GDPR Art. 26 and Regulation 2018/1725).
• Member State national competent authority: joint controller for national assessment and supervision (GDPR Art. 26).
• Sponsor: controller for trial conduct and participant data (GDPR Art. 4(7) and Art. 26).
• CRO, where appointed: processor (GDPR Art. 28).
Where a sponsor delegates processing activities to a Contract Research Organisation (CRO), a Data Processing Agreement (DPA) compliant with Article 28 of the GDPR must be in place. The JCA does not displace this bilateral obligation; it operates alongside it. For an overview of how these duties play out in practice, see our guidance on data protection for clinical trials.
Focus: Member State National Competent Authorities
National competent authorities (NCAs), such as the Medicines and Healthcare products Regulatory Agency (MHRA) in the United Kingdom (which operates under UK GDPR after Brexit), the Agence nationale de sécurité du médicament et des produits de santé (ANSM) in France and the Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) in Germany, act as joint controllers for personal data processed during the national assessment phase of a clinical trial application submitted via CTIS. Each NCA is responsible for ensuring that data submitted for national assessment is handled in accordance with the applicable national data protection law transposing or implementing the GDPR.
Focus: The Information Commissioner’s Office (ICO), United Kingdom
Following the United Kingdom’s departure from the European Union, CTIS is no longer accessible to UK based sponsors as a submission platform for UK trials. However, where a sponsor conducts a trial that spans both EU Member States and the United Kingdom, data flows between CTIS and UK trial sites will constitute international transfers subject to UK GDPR. The ICO’s International Data Transfer Agreement (IDTA) or the UK Addendum to the EU Standard Contractual Clauses may be required to legitimise such flows. Sponsors should assess this requirement at the protocol design stage rather than at the point of site activation. Our analysis of the data protection representative role in EU and UK clinical trials explores the related Article 27 obligations in depth.
Sponsor Obligations: From DPIA to Data Subject Rights
For sponsors, CTIS data protection compliance encompasses several distinct but interrelated obligations under the GDPR.
Data Protection Impact Assessment (DPIA): Article 35 of the GDPR requires a DPIA where processing is likely to result in a high risk to the rights and freedoms of natural persons. Clinical trials systematically process special category data within the meaning of Article 9(1) of the GDPR, specifically data concerning health. The WP29 Guidelines on Data Protection Impact Assessment (WP248 rev.01), endorsed by the EDPB, identify large scale processing of special category data as a criterion indicating high risk, which confirms that clinical research triggers the DPIA obligation. Sponsors must therefore complete a DPIA before CTIS submission and update it whenever the trial protocol is amended in a manner that materially alters the risk profile.
Legal basis: the appropriate legal basis for processing participant health data in the context of a clinical trial is typically Article 9(2)(j) of the GDPR (processing for scientific research purposes), read in conjunction with Article 6(1)(e) (performance of a task in the public interest) or Article 6(1)(b) (performance of a contract) depending on the sponsor’s legal form and the nature of the trial. Member States may impose additional conditions under Article 9(4).
Transparency and the ICF: the sponsor, as data controller, must provide participants with a privacy notice that satisfies Articles 13 and 14 of the GDPR. In practice, this information is typically embedded within the Informed Consent Form or provided as a standalone data protection information sheet. The JCA requires that sponsors make clear to participants which entities are processing their data, in what capacity and on what legal basis, a transparency obligation that becomes more complex where multiple joint controllers are involved.
Data minimisation: Article 5(1)(c) of the GDPR requires that personal data be adequate, relevant and limited to what is necessary in relation to the purposes for which it is processed. Within CTIS, this principle has practical implications for the data fields populated during submission: sponsors should populate only those fields that are strictly required for the assessment and supervision of the trial, avoiding the inclusion of unnecessary personal data concerning investigators or subinvestigators.
Data subject rights: under the JCA, sponsors bear primary responsibility for responding to data subject rights requests (access, rectification, erasure, restriction and portability under Articles 15 to 20 of the GDPR) in relation to participant data. Where a request relates to data held on the CTIS platform itself, the JCA establishes a coordination mechanism between EMA, the Commission and the relevant NCA. Sponsors should ensure that their trial level procedures dovetail with this mechanism and that timelines for response (one month, extendable to three months in complex cases under Article 12(3) of the GDPR) are embedded in their Standard Operating Procedures.
Focus Examples by Authority and Country
Focus: France, CNIL and MR-001
In France, sponsors conducting interventional clinical trials on medicinal products must comply with the Méthodologie de Référence MR-001 issued by the Commission Nationale de l’Informatique et des Libertés (CNIL). MR-001 constitutes a sector specific framework that provides a legal basis for processing health data in clinical research without the need for an individual authorisation from the CNIL, provided that the sponsor commits to complying with the methodology’s requirements. Compliance with MR-001 is documented through a commitment letter (lettre d’engagement) submitted to the CNIL. Where a trial does not satisfy all MR-001 conditions, for example because data will be transferred to a third country without adequate safeguards, the sponsor must seek an individual authorisation. CTIS submission does not replace MR-001 compliance; both obligations run concurrently. Our data protection strategies for Phase III clinical trials set out how to manage MR-001 alongside multi country requirements.
Focus: Germany, BDSG and State Level Requirements
In Germany, the Federal Data Protection Act (Bundesdatenschutzgesetz, BDSG) supplements the GDPR at national level. Section 27 BDSG provides a specific legal basis for processing personal data for scientific research purposes. Additionally, sponsors must engage with the relevant ethics committee (Ethikkommission) at state (Länder) level, as ethics committee oversight requirements vary across the sixteen Länder. BfArM, as the competent authority for the CTIS assessment phase in Germany, processes personal data in its capacity as a joint controller under the JCA. Sponsors should ensure that their DPIA accounts for the specific risk profile associated with processing under German law.
Cross Border Transfers and Third Country Considerations Within CTIS
CTIS is designed as an EU centric system, but the reality of global clinical development means that data submitted to CTIS will often originate from or be shared with entities in third countries. Sponsors headquartered outside the EEA, or those engaging CROs with operations in the United States, Japan, India or other jurisdictions, must ensure that any transfer of personal data from within the EEA to a third country complies with Chapter V of the GDPR.
The primary mechanisms available to sponsors are:
• Adequacy decisions under Article 45 of the GDPR (currently covering, among others, Japan, South Korea and the UK for standard personal data transfers).
• Standard Contractual Clauses (SCCs) adopted by the European Commission under Implementing Decision (EU) 2021/914, which must be supplemented by a Transfer Impact Assessment (TIA) to verify that the legal environment of the recipient country does not undermine the protection afforded by the SCCs.
• Binding Corporate Rules (BCRs) under Article 47 of the GDPR, applicable to intra group transfers.
Within the CTIS environment, sponsors should pay particular attention to the configuration of their electronic data capture (EDC) systems and trial master file (TMF) platforms. Where these systems are hosted by cloud service providers with infrastructure outside the EEA, a transfer mechanism and a TIA will be required before the system can be used to process data submitted to or derived from CTIS.
Focus: US Based Sponsors and the EU-U.S. Data Privacy Framework
US based pharmaceutical companies sponsoring trials with EU sites may benefit from the EU-U.S. Data Privacy Framework (DPF), established pursuant to Commission Implementing Decision (EU) 2023/1795 of 10 July 2023. Where a US sponsor or its CRO is certified under the DPF, personal data may be transferred from the EEA without the need for SCCs, provided that the transfer falls within the scope of the certification. Sponsors should verify DPF certification status at the outset of trial planning and monitor it throughout the trial, as certification must be renewed annually.
Key Concepts at a Glance
The following summary distils the core CTIS data protection concepts, the instruments that govern them and the action each requires from sponsors:
• Joint Controllership. Two or more controllers jointly determine the purposes and means of processing (GDPR Art. 26; JCA, effective 12 March 2026). Sponsor action: ensure the JCA is reflected in internal records and participant facing documentation.
• Data Processing Agreement. A contract governing a processor’s obligations (GDPR Art. 28). Sponsor action: execute a DPA with all CROs and service providers.
• DPIA. An assessment of high risk processing activities (GDPR Art. 35; EDPB Guidelines). Sponsor action: complete before CTIS submission and update on protocol amendments.
• Standard Contractual Clauses. A transfer mechanism for third country transfers (Commission Decision (EU) 2021/914). Sponsor action: execute alongside a Transfer Impact Assessment.
• MR-001 (France). A national sector methodology permitting processing without individual CNIL authorisation (CNIL Délibération). Sponsor action: submit a commitment letter and verify the conditions are met.
• Legal basis for research. Permits processing of health data for scientific research (GDPR Art. 9(2)(j) with Art. 6(1)(e) or (b)). Sponsor action: document in the DPIA and ICF.
• Data Subject Rights. Rights of access, rectification, erasure, restriction and portability (GDPR Arts. 15 to 20). Sponsor action: establish an SOP aligned with the JCA coordination mechanism.
How iliomad Can Support Your CTIS Compliance
CTIS data protection compliance demands specialist expertise at the intersection of EU clinical trials regulation and the GDPR. iliomad’s team of data protection and life sciences compliance specialists supports sponsors, CROs and investigator sites across the full lifecycle of a clinical trial, from protocol design and DPIA preparation through to audit support and data subject rights management. As regulatory compliance specialists rather than a law firm, iliomad focuses on operational implementation that stands up to inspection.
Our services include:
• Review and gap analysis of your JCA obligations and internal documentation.
• Drafting and negotiation of Data Processing Agreements with CROs and technology vendors.
• Transfer Impact Assessments for third country data flows.
• MR-001 commitment letters and CNIL interaction support.
• External Data Protection Officer cover for organisations required to appoint a Data Protection Officer under Article 37 of the GDPR, and EU Data Protection Representative services for sponsors established outside the EEA.
Ready to ensure your CTIS submissions are fully GDPR compliant? Contact the iliomad Clinical Trials Data Protection team to arrange an initial consultation.
FAQs
Our frequently questions
Yes. Clinical trials process special category health data on a large scale, which Article 35 of the GDPR treats as high risk, so a DPIA must be completed before CTIS submission. Update it whenever a protocol amendment materially changes the risk profile.
Controllership is shared. EMA, the European Commission and Member State national competent authorities act as joint controllers for platform and assessment activities under the Joint Controllership Arrangement, while the sponsor remains controller for the conduct of the trial and participant data. A CRO acting on the sponsor’s instructions is a processor.
No. The JCA governs the relationship between the CTIS joint controllers and runs alongside, not instead of, your bilateral obligations. You still need a Data Processing Agreement compliant with Article 28 of the GDPR with every CRO and vendor that processes data on your behalf.
Sometimes. If your organisation or your CRO is certified under the EU-U.S. Data Privacy Framework, personal data may flow from the EEA without SCCs, provided the transfer falls within the scope of the certification. Verify certification status at the planning stage and confirm it is renewed annually; otherwise use SCCs supported by a Transfer Impact Assessment.
After Brexit, CTIS is not a submission platform for UK trials, and data moving between CTIS and UK sites becomes an international transfer under UK GDPR. You will typically need the ICO’s International Data Transfer Agreement or the UK Addendum to the EU Standard Contractual Clauses. Assess this at protocol design rather than at site activation.
The sponsor takes primary responsibility for participant data subject rights requests under Articles 15 to 20 of the GDPR. Where the request concerns data held on the CTIS platform itself, the JCA establishes a coordination mechanism between EMA, the Commission and the relevant national competent authority. Build the response timelines (one month, extendable to three) into your Standard Operating Procedures.

Find out how iliomad can help your company.
Only visible in production

Weekly Digest: Healthcare Cyber Breaches, AI Regulation Advances and Genetic Data Enforcement | iliomad
Ransomware mortality data, the FDA-EMA AI framework, Germany's KI-MIG, the EU-US DPF under review and a wave of US healthcare breaches: this week's digest for biotech and healthtech.

EU Digital Omnibus Simplifies AI Act Obligations; Rhode Island Enacts AI Healthcare Laws
Regulation (EU) 2026/1744 streamlines EU AI Act compliance, while Rhode Island enacts three AI healthcare laws. Key updates for biotech and healthtech teams.

EDPB Anonymisation Guidelines 2026 and Clinical Trial Data: What Life Sciences Organisations Must Know
EDPB Guidelines 02/2026 on anonymisation set new standards for clinical trial data. Learn the three-criteria test, enforcement lessons and compliance steps. iliomad.
