



Summary
With the strict 2026 enforcement deadline fast approaching, does the uncertainty of achieving full EU AI Act ealthcare compliance leave your life sciences organization exposed to severe regulatory penalties and costly market exclusion? This technical analysis defines the specific high-risk classifications and mandatory governance frameworks you must implement immediately to align your medical software and devices with these uncompromising new European standards. You will discover a pragmatic, step-by-step roadmap designed to seamlessly integrate these rigorous data obligations into your existing quality management systems, ensuring your continued operation and safety oversight without disrupting clinical innovation.
1. High Risk Classification: Why Healthcare AI Is the Default Target
The Annex I and Annex III Reality
For life sciences companies pursuing EU AI Act healthcare compliance, the starting point is blunt: most AI applications in this sector are high risk by regulatory design, not by choice.
The EU AI Act classifies AI systems as high risk through two distinct pathways. Under Annex I, any AI system that is itself a medical device, or that functions as a safety component of one, requiring third party conformity assessment under the Medical Device Regulation (MDR) or In Vitro Diagnostic Regulation (IVDR) automatically qualifies. Under Annex III, AI systems used in healthcare access, triage, and clinical decision making are explicitly listed.
In practice, this captures a wide range of systems already deployed across biotech, pharma, and medtech: diagnostic imaging algorithms, clinical decision support systems (CDSS), patient triage tools, remote monitoring platforms, and AI driven trial recruitment software. Our work across 30+ life sciences clients, from clinical stage biotechs like MindMed and Formation Bio to medtech companies developing imaging solutions, consistently confirms that the high risk designation is the rule, not the exception.
Concrete Examples: What Falls Where
The classification is not always intuitive. Through our AI compliance work at Iliomad Health Data, we have mapped the most common life sciences AI use cases against the Act's risk tiers:
- High risk (Annex I, medical device pathway): AI powered radiology image interpretation, pathology slide analysis, dermatology lesion detection. These qualify as medical device software under MDR/IVDR and require third party conformity assessment.
- High risk (Annex III, healthcare access): AI based patient triage systems, clinical trial recruitment and matching platforms, symptom based prioritisation tools. These directly affect individuals' access to healthcare services.
- High risk (Annex III, biometrics): Biometric access control in laboratories, hospitals, and manufacturing facilities using facial recognition.
- Limited risk (transparency obligations): Patient facing chatbots handling scheduling or FAQs, subject to Article 50 disclosure requirements but not the full high risk regime.
- Minimal risk: AI used solely for internal R&D (molecule screening, protein interaction prediction) or back office productivity (document review, contract analysis). Subject primarily to AI literacy obligations under Article 4.
One critical nuance: AI for clinical trial monitoring and site performance analytics may start as limited risk but escalates to high risk the moment its outputs influence participant safety decisions or trial continuation. Context of use is decisive.
The MDR/IVDR Layer: Additive, Not Substitutive
The AI Act does not replace MDR or IVDR. It stacks on top. When an AI system functions as a medical device, both regulatory frameworks must be satisfied simultaneously. Compliance with one does not discharge obligations under the other.
This creates a dual conformity burden: technical documentation, risk management systems, and quality management systems (QMS) must rigorously address the specific demands of both legal regimes. A single conformity assessment pathway exists only where the AI Act requirements are fully integrated into the notified body assessment under the relevant MDR/IVDR procedure.
2. What High Risk Providers Must Deliver
Once a system carries the high risk classification, a defined set of obligations applies before any market placement. These are not aspirational; they are legally enforceable prerequisites.
Quality and Risk Management: Lifecycle Coverage
A Quality Management System is required that governs every phase of the AI system's lifecycle, from design and data preparation through deployment and decommissioning. Organisations already operating under ISO 13485 for medical devices should layer the AI Act requirements into that existing framework rather than build a parallel silo.
The risk management system must specifically identify, evaluate, and mitigate AI specific hazards: algorithmic bias, data drift, adversarial inputs, and failure modes in clinical environments. This goes beyond traditional medical device risk management under ISO 14971 by requiring continuous post deployment risk monitoring.
Data Governance: Where Most Will Fail
The AI Act's data governance requirements under Article 10 are among the most operationally demanding provisions. Training, validation, and testing datasets must be demonstrably relevant, representative, and free from errors that could introduce bias into medical outcomes.
For life sciences companies running international clinical trials, this intersects directly with existing data protection compliance obligations. The datasets feeding algorithms must satisfy both the AI Act's quality standards and GDPR's lawfulness requirements, including lawful basis for processing, data minimisation, and cross border transfer safeguards.
Every step of the data governance lifecycle must be documented. This is not a one time exercise but a living record that regulators can audit at any point.
Transparency, Explainability, and Human Oversight
Black box algorithms are a regulatory liability under the AI Act. Providers must ensure that users, including clinicians, hospital IT teams, and clinical research associates, can meaningfully interpret outputs and understand the system's capabilities and limitations.
The core operational requirements are:
- Risk management system: Maintained throughout the entire AI lifecycle, not just at deployment.
- Data quality assurance: Documented protocols ensuring training data is relevant, representative, and bias tested.
- Clear instructions for use: Clinicians must understand what the AI output means and when to override it.
- Human oversight by design: The system must allow effective human supervision, intervention, and shutdown.
Self declaration of conformity is rarely available for healthcare AI. Most systems will require a third party conformity assessment through a Notified Body. The penalty for non compliance is severe: financial sanctions up to €35 million or 7% of global annual turnover, and prohibition on marketing the product in the EU.
3. The Compliance Timeline: Three Deadlines That Define the Strategy
The AI Act's deployment is staggered. Each phase carries binding obligations.
February 2025: Prohibitions in Force
The first wave banned AI practices classified as unacceptable risk: social scoring, subliminal manipulation, exploitation of vulnerabilities. While these rarely apply directly to clinical operations, they set the enforcement tone. Ignoring early compliance signals risks broader regulatory scrutiny.
August 2025: GPAI and Governance Rules
Obligations for General Purpose AI (GPAI) models and national governance structures became effective in August 2025. Life sciences companies that build on foundation models from providers like OpenAI, Google, or Anthropic face new transparency and documentation requirements as downstream deployers. The GPAI provider's compliance cannot be assumed to cover a specific downstream use case.
August 2026: Full High Risk Regime
This is the definitive deadline. Every requirement for high risk AI systems, including conformity assessment, technical documentation, post market monitoring, and incident reporting, becomes fully enforceable. Any high risk system on the EU market after this date must be entirely compliant.
Retrofitting compliance after August 2026 is not a viable strategy. The conformity assessment process alone requires months of preparation. Companies that delay preparation until early 2026 will face bottlenecks with Notified Bodies and risk market exclusion.
4. The Regulatory Stack: AI Act, MDR/IVDR, EHDS, PLD, and GDPR
EU AI Act healthcare compliance does not exist in isolation. It sits within an interconnected web of regulations that life sciences companies must navigate as a unified framework.
European Health Data Space (EHDS): Data Access Under Conditions
The EHDS creates a regulated framework for secondary use of electronic health data, including for AI algorithm training and validation. For companies struggling to source the high quality datasets demanded by Article 10 of the AI Act, the EHDS is a strategic opportunity.
However, access is conditional. Data must be accessed through authorised Health Data Access Bodies, and processing must comply with GDPR safeguards. The EHDS solves the data availability problem but does not waive data protection obligations.
Product Liability Directive (PLD): Software as Product
The revised PLD explicitly classifies software, including AI systems, as products. AI providers now face the same strict liability regime as physical manufacturers. When a system fails to comply with EU safety requirements, courts may presume it defective. Non compliance with the AI Act effectively guarantees liability exposure.
GDPR: The Permanent Foundation
The GDPR remains the bedrock. Any personal data used for AI training, validation, or operational processing must comply with its principles: lawful basis, data minimisation, purpose limitation, and individual rights. For companies running multi jurisdictional clinical trials, which is the core of our client base at Iliomad, the complexity of maintaining simultaneous compliance with GDPR, the AI Act, and local data protection laws across 70+ countries requires a structured, jurisdiction by jurisdiction approach.
5. Deployer Obligations: What Hospitals and Clinics Owe
Compliance responsibility does not stop with the provider. Healthcare institutions deploying high risk AI carry their own distinct obligations.
Fundamental Rights Impact Assessment (FRIA)
Public bodies and entities providing public services must conduct a Fundamental Rights Impact Assessment before deploying high risk AI. This requires mapping how the system might affect patient rights, identifying specific risks of harm, and defining governance measures to mitigate them.
AI Literacy and Human Oversight
Article 4 mandates that deployer staff possess sufficient AI literacy to operate the system responsibly. Clinicians must understand what the tool can and cannot do. They must have the authority to override or stop the system at any time.
Deployer specific obligations include:
- Staff training: Clinical and IT personnel must be trained on the specific AI system's capabilities, limitations, and failure modes.
- Input data governance: The deployer must confirm that real world input data is relevant to the system's intended purpose and validated environment.
- Incident reporting: Serious incidents or identified risks must be reported promptly to the provider and, where required, to the relevant market surveillance authority.
Post Deployment Monitoring
Deployers must monitor the AI system's performance in real world clinical conditions, detect drift from intended use, and maintain automatically generated logs as evidence of compliance. This operational monitoring is a continuous obligation, not a one time assessment.
6. The 2026 Governance Roadmap: Integrate, Don't Duplicate
Merge AI Act Requirements Into the Existing QMS
The most effective approach for life sciences companies is integration, not duplication. AI Act obligations covering data governance, risk management, and post market monitoring belong inside the existing Quality Management System alongside MDR/IVDR technical documentation. Building a parallel compliance silo creates confusion, increases cost, and weakens audit readiness.
The goal is a single conformity assessment pathway with unified technical documentation that satisfies both the medical device and AI regulatory regimes.
Build the Data Governance Strategy Now
Organisations must evaluate data collection, curation, and management processes immediately. Documented protocols are required that guarantee relevance, representativeness, and absence of bias in every dataset feeding AI systems. Under Article 10, this documentation is non negotiable for regulatory approval.
Consider Appointing an AI Compliance Officer
While the AI Act does not mandate the appointment of an AI Officer, organisations that establish this role position themselves with a dedicated compliance leader: an internal watchdog who oversees conformity assessments, supervises the risk management framework, maintains technical documentation, and serves as the liaison with market surveillance authorities.
At Iliomad Health Data, we offer this function through two distinct service units: AI Governance & Compliance Services (acting as AI Compliance Officer) and AI Regulatory Representation Services (acting as EU Authorised Representative under Article 22). These units operate with separate reporting lines and independent review processes, because the entity creating compliance documentation must never be the same one verifying it.
Actionable Steps
- Conduct a gap analysis of all current and planned AI systems against Annex I, Annex III, and the full high risk requirements.
- Map the regulatory stack: Identify where AI Act obligations overlap with existing MDR/IVDR, GDPR, and cybersecurity frameworks.
- Assign governance ownership: Designate a specific person or team, internal or external, responsible for AI compliance oversight.
- Engage Notified Bodies early: Conformity assessment capacity is limited. Starting discussions now avoids bottlenecks in 2026.
- Implement post market surveillance: Define how deployed systems will be monitored, integrating AI performance monitoring with existing vigilance and post market data protection obligations.
- Invest in AI literacy training: Ensure clinical, quality, regulatory, and IT teams understand the AI systems they develop, deploy, or supervise.
FAQs
Our frequently questions
Most AI systems intended for medical purposes qualify as high risk through one of two pathways. Under Annex I, AI that functions as a medical device or safety component requiring third party conformity assessment under MDR/IVDR is automatically high risk. Under Annex III, systems affecting healthcare access, triage, clinical decisions, or biometric identification are explicitly listed. This captures diagnostic imaging, clinical decision support, patient monitoring, and trial recruitment tools. Internal R&D tools (drug discovery, molecule screening) are generally minimal risk, and patient facing chatbots without clinical functions carry transparency obligations only.
The AI Act applies as an additional regulatory layer, not a replacement. AI systems that qualify as medical devices must satisfy both frameworks simultaneously. The most efficient approach is integrating AI Act requirements, particularly data governance, transparency, and fundamental rights obligations, into the existing QMS and technical documentation under ISO 13485. Where feasible, a single conformity assessment can address both regimes, but the technical documentation must explicitly cover each framework's specific demands.
The full high risk regime becomes enforceable in August 2026. However, the timeline is staggered: prohibited practices have applied since February 2025, and GPAI rules took effect in August 2025. The transition period between now and August 2026 is not a grace period but rather the preparation window. Companies that start conformity assessment preparation in mid 2026 will face Notified Body capacity constraints and risk market exclusion.
The revised PLD classifies software, including AI systems, as products subject to strict liability. Providers can be held liable for damages without the claimant proving negligence. When an AI system fails to comply with mandatory EU safety requirements, including the AI Act, it may be presumed defective. This presumption significantly increases legal exposure for non compliant providers.
Yes. Deployers must ensure human oversight, monitor system performance in real world conditions, maintain operational logs, and report serious incidents. Public sector deployers must also conduct a Fundamental Rights Impact Assessment (FRIA) before deploying high risk systems. Article 4 additionally requires deployer staff to possess sufficient AI literacy to operate the system safely and interpret its outputs.
The AI Act demands high quality, representative, error free training data. The EHDS provides a regulated framework for accessing electronic health data for secondary uses, including AI development and validation. It addresses the data availability challenge but operates under strict conditions: access must go through Health Data Access Bodies and comply fully with GDPR. The EHDS is a data sourcing mechanism, not a compliance shortcut.
An AI Compliance Officer oversees conformity assessments, supervises risk management, maintains technical documentation, and acts as the liaison with regulators. While not legally mandated by the AI Act, the role mirrors the DPO function under GDPR and provides organisations with dedicated compliance leadership. For life sciences companies, this function is particularly valuable given the intersection of AI, medical device, and data protection regulations, three domains that require coordinated governance.

Find out how iliomad can help your company.
Only visible in production

Data Protection in Clinical Trials: Governing AI-Assisted Data Flows Under GDPR
AI intensifies clinical trial data protection, requiring end-to-end governance and inspection-ready controls.
Weekly Digest: EU AI Act Prohibitions, GDPR Enforcement and Clinical Trial Developments - Week of 22 June 2026
AI healthcare rules tighten as EU, US and GDPR developments accelerate.
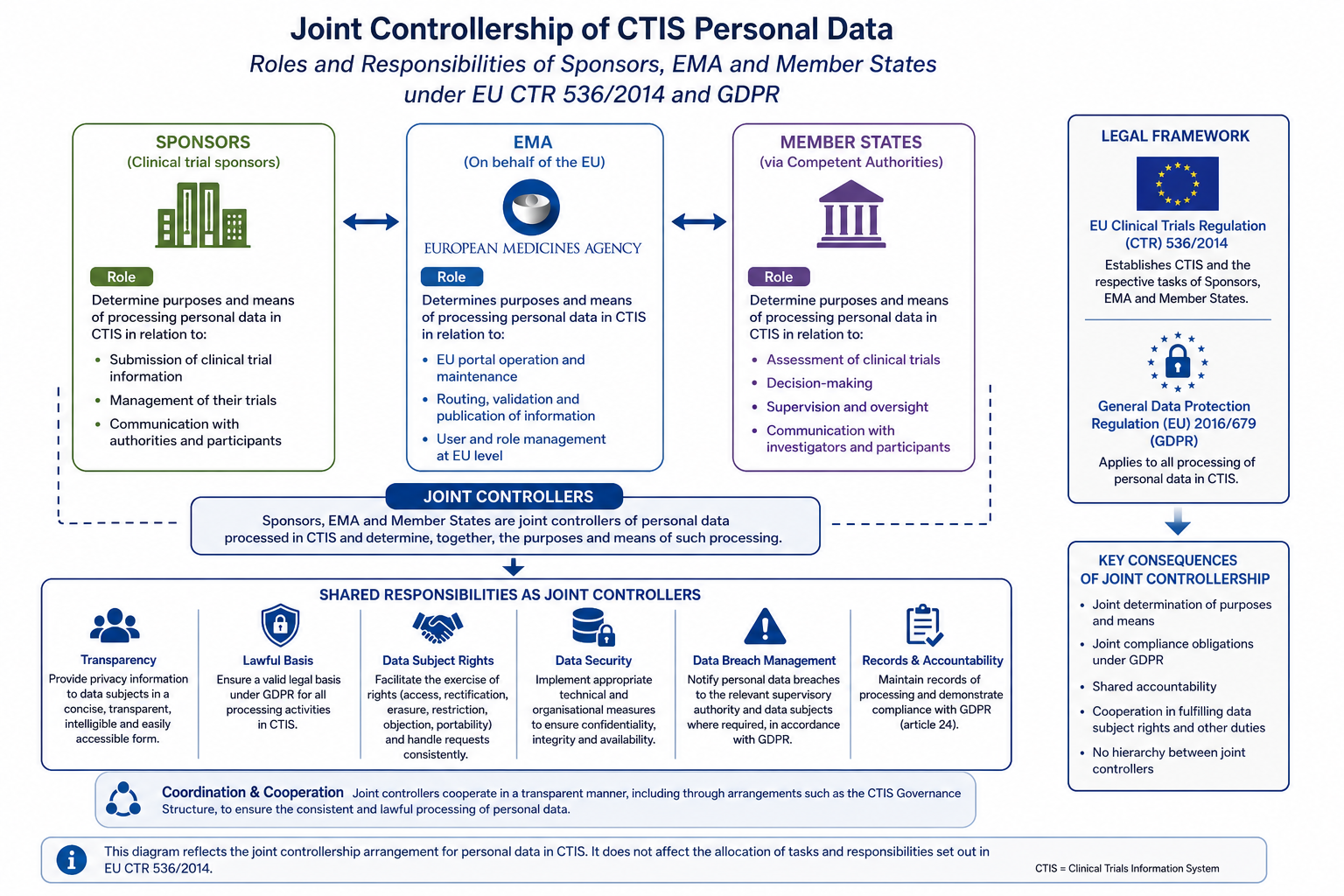
CTIS Data Protection: Joint Controllership, GDPR Obligations and Sponsor Responsibilities
Understand CTIS data protection obligations under EU CTR 536/2014 and GDPR. Learn how the Joint Controllership Arrangement allocates roles among sponsors, EMA and Member States.
