



Summary
This article guides EU health tech companies entering the US market through the multi-layered regulatory framework for AI medical devices, covering FDA oversight, HIPAA privacy rules, state laws, and IRB approval processes. It details three consent exception mechanisms (minimal risk waiver, life-threatening exception, emergency research exception) for research involving incapacitated patients, explaining why minimal risk waivers are often most practical for AI diagnostic tool validation. The guide also addresses the critical question of secondary data use for AI model training, emphasizing that companies must plan for this from day one by including model improvement purposes in original protocols, consents, and IRB submissions.
A significant trend is emerging in the life sciences and health technology sectors: European companies are increasingly looking to the United States as their primary market forAI-powered medical devices, sometimes bypassing the EU market entirely. This strategic shift reflects the complexities of European regulatory frameworks and the substantial opportunities presented by the American healthcare system.
For companies developing artificial intelligence tools in healthcare—particularly those involving diagnostic imaging, patient assessment, or clinical decision support understanding the intricate web of US regulations is essential. Unlike the European Union’s relatively harmonised approach under the Medical DeviceRegulation (MDR) and General Data Protection Regulation (GDPR), the UnitedStates presents a multi-layered regulatory environment that includes federal agencies, state-specific laws, and institution-level requirements.
This comprehensive guide draws on practical experience helping health technology companies navigate these complexities, offering actionable insights for organisations seeking to validate and deploy AI-powered medical devices in the US market. We examine the regulatory landscape from multiple angles: the FDA’s oversight of software as a medical device, HIPAA’s privacy requirements, state-level data protection laws, and the critical role of Institutional Review Boards in approving research involving human subjects.
Understanding the US Regulatory Landscape: A Multi-Layered System
The Department of Health and Human Services Ecosystem
The United States Department ofHealth and Human Services (HHS) serves as the umbrella organisation overseeing healthcare regulation in America. Within this structure, several agencies play distinct but interconnected roles in regulating AI-powered medical devices and the research required to validate them.
The Food and DrugAdministration (FDA) holds primary responsibility for ensuring the safety and effectiveness of medical devices, including software applications that qualify as medical devices. The FDA provides guidelines that apply both to research activities (clinical investigations) and to the commercial deployment of approved devices.
The Office for Human ResearchProtections (OHRP) oversees research involving human subjects, establishing requirements designed to protect research participants. When a company needs to conduct clinical investigations to validate an AI tool, OHRP’s regulations come into play alongside FDA requirements.
Institutional Review Boards(IRBs) represent the ground-level implementation of federal research protections. These bodies review and approve research protocols, ensuring that studies meet ethical standards and regulatory requirements. Understanding howIRBs operate is crucial for any company planning clinical research in theUnited States.
The IRB System: Key Differences from European Ethics Committees
For companies familiar withEuropean ethics committees, the American IRB system presents both similarities and important differences that can significantly impact project timelines and compliance strategies.
In Europe, ethics committees typically operate at the regional level, with study dossiers assigned to committees through centralised systems. The United States takes a different approach: IRBs can exist at multiple levels, including within individual healthcare institutions. A major university hospital, for example, will have its own IRB with specific guidelines and requirements.
This institutional structure means that requirements can vary significantly between IRBs, even within the same state. One IRB might approve a protocol that another rejects, creating potential inconsistencies that sponsors must navigate carefully. When selecting research sites, understanding the local IRB’s historical positions and requirements can help avoid unexpected obstacles.
Several additional features distinguish the US IRB system:
Private IRBs: UnlikeEurope’s primarily public ethics committee system, the United States has commercial IRBs that offer expedited review services for a fee. Companies seeking faster turnaround times can engage these private IRBs, though this approach requires careful consideration of costs and credibility factors.
Central IRB Mechanisms: For multi-site studies, a centralised IRB approach allows sponsors to submit protocols to a single IRB that then coordinates with individual sites. This mechanism can significantly streamline the approval process for studies spanning multiple hospitals or research centres across different states.
Software as a Medical Device: FDA Classification and Requirements
Understanding SaMD Classification
The FDA’s framework for regulating Software as a Medical Device (SaMD) has evolved significantly in recent years as artificial intelligence and machine learning applications have proliferated in healthcare. For companies developing AI-powered diagnostic or monitoring tools, understanding how the FDA classifies and regulates SaMD is foundational to market entry strategy.
SaMD refers to software intended to be used for medical purposes without being part of a hardware medical device. An AI algorithm that analyses patient images to detect clinical ab normalities, for example, would typically qualify as SaMD because it performs a medical function (diagnosis) independent of any specific hardware platform.
The FDA classifies SaMD based on the significance of the information provided by the software to healthcare decisions and the state of the healthcare situation. Tools that inform clinical management of serious or critical conditions face more stringent regulatory requirements than those providing low-risk wellness information.
The Validation Imperative
Before an AI-powered medical device can be legally marketed and deployed in the United States, it must be validated through clinical investigation. This requirement creates a chicken-and-egg challenge: the company needs data to validate its tool, but collecting that data involves regulatory requirements that assume the tool’s purpose is already established.
Structuring the validation pathway correctly from the outset is essential. Companies must clearly distinguish between:
Phase One: Research andValidation — During this phase, the company conducts clinical investigations to generate evidence supporting the safety and effectiveness of its AI tool. Data collection occurs under research regulations, with specific consent and IRB approval requirements.
Phase Two: CommercialDeployment — Once FDA authorisation is obtained, the company can deploy itstool commercially. Data collection during this phase occurs under different regulatory frameworks, primarily HIPAA and state laws governing treatment and diagnosis activities.
This distinction matters enormously for compliance planning. The regulations governing data collection, consent requirements, and permissible uses differ significantly between research and commercial deployment contexts. Companies that conflate these phases often encounter regulatory obstacles that could have been avoided with proper structuring.
HIPAA in the Research Context: Applicability and Exceptions
When Does HIPAA Apply to AI Medical Device Research?
The Health Insurance Portability and Accountability Act (HIPAA) Privacy Rule establishes national standards for protecting individuals’ health information. For companies developing AI medical devices, understanding HIPAA’s applicability—and its limits—is crucial for designing compliant data collection strategies.
A common misconception among health technology companies is that HIPAA applies directly to them as device developers. In most research contexts, this is not the case. HIPAA’s PrivacyRule applies to “covered entities”—primarily healthcare providers, health plans, and healthcare clearinghouses—and their “business associates.”
A company conducting research to validate an AI medical device typically does not qualify as a covered entity.The company is not providing healthcare; it is developing a tool that healthcare providers may eventually use. This distinction has important practical implications.
However, HIPAA nonetheless affects AI medical device research because the data needed for validation must be obtained from covered entities. Hospitals, clinics, and other healthcare organisations that collect patient data are covered entities. When these organisations share protected health information (PHI) with a device developer for research purposes, HIPAA governs what they can share and under what conditions.
This means that while the device developer may not be directly subject to HIPAA, the practical effect is similar: data can only flow to the developer if HIPAA’s requirements for research disclosures are satisfied. Understanding these requirements is essential for designing feasible data collection protocols.
The De-Identification Question
One potential pathway aroundHIPAA’s restrictions is de-identification. If data is properly de-identified under HIPAA standards, it is no longer considered PHI and can be used without the restrictions that apply to identifiable health information.
HIPAA establishes specific standards for de-identification, including the “Safe Harbor” method that requires removal of eighteen specific identifiers. For many AI applications, this approach is feasible and attractive—de-identified data can be used freely for algorithm training and validation.
However, for AI tools that analyse images, video recordings, or biometric data, de-identification presents fundamental challenges. HIPAA’s eighteen identifiers include full-face photographs, comparable images, and biometric identifiers including voice prints.
For an AI tool designed to analyse visual or biometric features to detect medical conditions, these identifiers cannot be removed without destroying the data’s utility. The very features the algorithm needs to analyse are the features that make the data identifiable under HIPAA.
This reality means that companies developing image-based or biometric AI diagnostic tools generally cannot rely on de-identification to avoid HIPAA restrictions. Alternative compliance pathways—particularly consent and waiver mechanisms—become essential.
HIPAA Authorisation and Waiver Mechanisms
When de-identification is not feasible, HIPAA provides two primary mechanisms for research use of PHI:individual authorisation and IRB waiver.
Individual Authorisation: Under normal circumstances, covered entities may disclose PHI for research purposes only with the individual’s written authorisation. This authorisation must meet specific requirements, including a description of the information to be disclosed, identification of recipients, and an expiration date or event.
For many research contexts, obtaining individual authorisation is straightforward. Participants in a clinical trial, for example, typically sign authorisation forms along withtheir informed consent documents.
IRB Waiver of Authorisation: HIPAA recognises that some research cannot practically be conducted if individual authorisation is required. The Privacy Rule therefore allows IRBs (or PrivacyBoards) to waive the authorisation requirement under specific conditions.
To grant a waiver, the IRB must find that:
- The use or disclosure involves no more than minimal risk to individuals’ privacy
- The research could not practicably be conducted without the waiver
- The research could not practicably be conducted without access to the PHI
For AI medical device research involving patients who cannot provide consent—for instance due to their clinical condition—the waiver mechanism may be the only viable path.Understanding how to present a compelling case for waiver to an IRB is a critical skill for companies in this space. The application must demonstrate that privacy risks are minimised, that the research serves important purposes, and that alternatives to using identifiable data have been considered and found inadequate.
Consent Waivers for Research Involving Subjects Unable to Consent
The Challenge of Incapacitated Research Subjects
One of the most complex regulatory challenges in AI medical device development involves research with subjects who cannot provide informed consent. This situation arises in various clinical contexts where patients may be unconscious, cognitively impaired, sedated, or otherwise unable to understand and agree to research participation.
For AI tools designed to support clinical decision-making in acute care settings, this challenge may be inherent to the use case. The tool may be most valuable precisely when patients are experiencing symptoms that impair their capacity to consent. Requiring consent before data collection could either exclude the most clinically relevant patient population or delay data collection until after the critical diagnostic window has passed.
US regulations recognise this challenge and provide several exception mechanisms. Understanding which exception applies—and the conditions for its use—is essential for structuring compliant research protocols.
Three Categories of Consent Exceptions
Federal regulations establish three primary categories of exceptions to informed consent requirements for research:
Minimal Risk Waiver (21 CFR 50.22 equivalent provisions)
This exception allows IRBs to waive consent requirements for research that presents no more than minimal risk to subjects. The concept of “minimal risk” is defined as risk not greater than that encountered in daily life or during routine medical examinations.
For AI diagnostic tool validation, the minimal risk waiver may be applicable when the research involves only observation and data collection, without any intervention that could harm the patient. If the AI tool is analysing data that would be captured anyway as part of standard care, and the research adds no additional procedures or risks, the minimal risk standard may be satisfied.
The minimal risk waiver requiresIRB approval and documentation of findings supporting the waiver. It cannot be self-determined by the research sponsor.
Life-Threatening Exception (21 CFR 50.23)
This exception permits research without consent on an ad hoc, individual basis when obtaining consent is not feasible due to the subject’s condition. Unlike a blanket waiver, this exception operates case-by-case: for each subject, the investigator must determine that consent cannot be obtained before proceeding without it.
The life-threatening exception requires that:
- The subject faces a life-threatening situation requiring immediate intervention
- Available treatments are unproven or unsatisfactory
- Obtaining consent is not feasible due to the subject’s condition
- The research holds prospect of direct benefit to the subject
This exception is narrower than it might initially appear. It is designed primarily for research involving experimental treatments where the intervention itself might benefit the subject. For purely observational research or research involving devices that are still being validated, the “prospect of direct benefit” requirement may not be satisfied.
Exception from Informed Consent for Research in Certain Acute Conditions(21 CFR 50.24)
This exception is the broadest but also the most procedurally demanding. It allows research without consent when the research involves subjects who cannot consent due to their medical condition, the condition is life-threatening, and the research must be conducted before consent can be obtained.
Importantly, this exception applies prospectively to all subjects meeting the criteria, not on a case-by-case basis. This makes it suitable for research involving acute clinical conditions where subject incapacity is expected.
However, the exception comes with extensive requirements:
- Community consultation before the research begins
- Public disclosure of the research plan
- Establishment of an independent data monitoring committee
- Protocols for attempting to contact family members
- Procedures for providing information to subjects after the fact
These requirements make this exception resource-intensive to implement. For many AI medical device validation studies, the minimal risk waiver—if applicable—provides a more practical path.
Practical Guidance on Exception Selection
Choosing the appropriate consent exception requires careful analysis of the specific research protocol and patient population. Several considerations inform this choice:
Patient Capacity Spectrum: Not all patients experiencing the target condition will be incapacitated. The patient population may range from fully alert individuals with mild symptoms to unconscious patients with severe presentations. If a substantial portion of the target population can consent, a blanket exception may not be appropriate.
Risk–Benefit Analysis: Purely observational research—where the AI tool is not influencing treatment decisions during the validation phase—presents different risk profiles than interventional research. Lower-risk research is more likely to qualify for minimal risk waivers.
IRB Expectations: DifferentIRBs may have different perspectives on exception applicability. Understanding the specific IRB’s historical positions and requirements helps in designing protocols likely to receive approval.
For many AI medical device validation studies, the recommended approach is to:
- Design the protocol to minimise risk, keeping data collection observational where possible
- Apply for minimal risk waiver as the primary exception mechanism
- Include provisions for obtaining consent from capable subjects
- Document the analysis supporting exception selection for IRB review
HIPAA in the Commercial Deployment Context
The Business Associate Relationship
Once an AI medical device receives FDA authorisation and enters commercial deployment, the regulatory framework shifts. The company is no longer conducting research; it is providing a service to healthcare providers. This shift changes HIPAA’s applicability.
In the commercial context, an AI medical device company may qualify as a “business associate” under HIPAA. A business associate is a person or organisation that performs functions or activities on behalf of a covered entity that involve access to PHI.
An AI tool that receives patientdata from a hospital, analyses it to detect clinical indicators, and returns results to the clinical team is performing a function involving PHI access. The company providing this tool would typically be considered a business associateof the hospital.
Business associate status triggers several requirements:
- A formal Business Associate Agreement (BAA) must be executed with each covered entity client
- The business associate must comply with HIPAA SecurityRule requirements
- The business associate faces direct liability for HIPAA violations
- Use and disclosure of PHI is limited to the purposes specified in the BAA
The Treatment Exception in Commercial Deployment
A crucial distinction between research and commercial deployment involves HIPAA’s treatment exception. UnderHIPAA, covered entities may use and disclose PHI for treatment purposes without individual authorisation. A hospital can share patient information with specialists, laboratories, and other providers involved in the patient’s care without obtaining specific consent for each disclosure.
When an AI medical device is deployed commercially as part of clinical care, the treatment exception may apply. If the device is being used to diagnose or inform treatment decisions for the patient whose data is being analysed, the disclosure from hospital to device provider can occur under the treatment exception.
This represents a significant practical advantage for commercial deployment compared to research. During the research phase, specific authorisations or waivers were required because the purpose was validation, not treatment. During commercial deployment, if the purpose is treatment, the authorisation requirement falls away.
However, the treatment exception has limits. It covers use and disclosure of PHI for the treatment of the individual whose information is at issue. It does not cover uses that benefit other patients or the healthcare system generally.
Secondary Use of Data for AI Model Training and Improvement
The Critical Question of Model Refinement
One of the most important regulatory questions for AI medical device companies concerns secondary use of data. After data has been collected—whether during research or commercial deployment—can it be used to train, refine, and improve the AI model?
This question matters enormously for AI development. Machine learning models improve with more data. A company that can use clinical data from deployed devices to refine its algorithms gains significant competitive and clinical advantages. But regulatory compliance isn ot automatic.
The answer depends on several factors: the context in which data was originally collected, the consents or authorisations obtained, and the specific use contemplated.
Secondary Use in the Research Context
When data is collected during a clinical investigation, the permissible uses are defined by the research protocol and consent/authorisation documents. If the protocol and authorisations specifically include AI model training as a purpose, secondary use is permitted within those bounds.
This is why careful protocol design is essential. Companies should consider secondary use possibilities at the protocol drafting stage and ensure that:
- The research protocol explicitly describes AI model training as a study purpose
- IRB approval covers this purpose
- Any consent forms or authorisation documents reference model training
- HIPAA waivers (if applicable) encompass the secondary use
If secondary use was notcontemplated in the original protocol, options are limited. The company can seek amended IRB approval and additional HIPAA waiver for the secondary purpose, obtain fresh consent/authorisation from subjects (often impractical),or de-identify the data to HIPAA standards (often not feasible for image-basedAI). The lesson is clear: think ahead. Including secondary use provisions fromthe outset is far easier than seeking retroactive authorisation.
Secondary Use in the Commercial Deployment Context
When data is collected during commercial deployment under the treatment exception, secondary use for model training presents different challenges. The treatment exception permits use and disclosure for treatment purposes. Model training—which benefits future patients and the company, not the individual whose data is used—is not treatment.
This means that data collected under the treatment exception cannot automatically be used for model training.The company must find another legal basis for this secondary use.
Options include:
Obtaining Explicit Authorisation: The company can request that patients authorise use of their data for model training. This requires developing authorisation forms that meetHIPAA standards and implementing processes to obtain signatures. For some clinical settings this may be impractical during the acute care episode but could be pursued during follow-up.
HIPAA Research Waiver: If model training qualifies as research, the company can seek an IRB waiver of authorisation. This requires establishing a research protocol, securing IRB review, and demonstrating that waiver criteria are met. This approach reframes commercial model training as a research activity subject to research regulations.
De-Identification: If data can be de-identified to HIPAA standards, it exits the HIPAA regulatory framework and can be used freely. For AI applications that can function with de-identified data, this path is attractive. For image-based or biometric applications, it is typically unavailable.
State Law Considerations for Secondary Use
Beyond HIPAA, state privacy laws may impose additional restrictions on secondary use. California and Texas, for example, have specific requirements regarding use of health information that may exceed HIPAA’s baseline.
However, state laws often provide more flexible approaches to de-identification than HIPAA’s rigid eighteen-identifier standard. Under many state laws, data is considered de-identified if it cannot reasonably be used to identify an individual, without requiring removal of specific categories of information.
This flexibility may create opportunities. A company might find that data which cannot be de-identified under HIPAA’s Safe Harbor standard can nonetheless be de-identified under state law through techniques like removing metadata and demographic information, blurring or obscuring identifying features in images, applying differential privacy techniques, or aggregating data to prevent individual identification.
The analysis must be jurisdiction-specific. What constitutes adequate de-identification varies between states and between HIPAA and state standards. Companies operating across multiple states need compliance strategies that account for this variation.
Practical Recommendations for AI Medical Device Companies
Structure the Problem Before Solving It
Companies entering the US healthcare market often arrive with a clear vision of their technology but a vague understanding of the regulatory pathway. The first step in any compliance engagement should be structuring the problem.
Key structuring questions include:
- Is the immediate need research (validation) or commercial deployment?
- Which federal agencies have jurisdiction over the intended activities?
- Which states are targeted, and what state-specific requirements apply?
- What is the realistic patient population, and what capacity issues arise?
- What data elements are essential, and can any bede-identified?
Taking time to answer these questions before diving into regulatory details prevents wasted effort and ensures that compliance strategies align with business realities.
Anticipate Secondary Use from Day One
The importance of planning for secondary use cannot be overstated. Adding model training provisions to an existing protocol or consent is possible but cumbersome. Including these provisions from the start is straightforward.
Every research protocol should explicitly address whether collected data may be used for training and improving the AI model under development, developing future AI models or applications, sharing with research collaborators, and publication in de-identified or aggregated form.
Every consent form and HIPAA authorisation should address these uses in language that subjects can understand. And every IRB submission should explain the secondary use rationale and seek approval for the full scope of intended uses.
Build Relationships with IRBs Early
IRB approval is often on the critical path for US market entry. Building relationships with relevant IRBs before formal submissions can smooth the approval process.
Consider requesting pre-submission meetings to discuss the protocol concept, understanding the specific IRB’s perspectives on consent exceptions, identifying any unique local requirements or preferences, and establishing communication channels with IRB staff.
For multi-site studies, evaluating central IRB options early can simplify coordination. A single central IRB can provide approvals that cover multiple research sites, reducing the administrative burden of site-by-site submissions.
Document Everything for Future Reference
US regulatory compliance generates substantial documentation: protocols, consent forms, IRB approvals,HIPAA analyses, FDA submissions. This documentation should be organised and preserved for future reference.
Beyond immediate compliance needs, this documentation supports future regulatory submissions building on prior work, defence against any compliance inquiries or audits, training for new team members, and expansion to new jurisdictions or use cases. Investing in documentation management early prevents scrambling to reconstruct record slater.
Engage Specialised Expertise
The US regulatory landscape forAI medical devices is complex, evolving, and high-stakes. Errors can delay market entry by months or years, or expose companies to enforcement actions and liability.
Engaging advisors with specific expertise in FDA regulation, HIPAA compliance, and IRB processes is a sound investment. Look for expertise that combines deep knowledge of applicable regulations, practical experience with similar technologies and use cases, understanding of how agencies and IRBs actually operate, and the ability to translate regulatory requirements into operational guidance.
The goal is not just knowing the rules but understanding how to apply them effectively to achieve business objectives while maintaining compliance.
Conclusion: Complexity That Rewards Careful Navigation
The US regulatory environment for AI medical devices is undeniably complex. Multiple federal agencies, state-specific laws, institution-level requirements, and evolving guidance onAI create a landscape that can overwhelm companies accustomed to more streamlined regulatory systems.
Yet this complexity is navigable. Companies that invest in understanding the regulatory framework, structure their approach thoughtfully, and engage appropriate expertise consistently achieve successful US market entry. The key is recognising that compliance is not a checkbox exercise but a strategic discipline that shapes product development, clinical validation, and commercial deployment.
For European companies considering the US market—a trend that shows no signs of abating—the message is clear. The opportunities are real, but so are the regulatory requirements.Success comes to those who take compliance seriously from the outset, building regulatory strategy into product strategy rather than treating it as an afterthought.
The companies that thrive in this environment share common characteristics: they understand their regulatory obligations, they plan ahead for secondary use and future applications, they build relationships with regulatory bodies and IRBs, and they document their compliance efforts thoroughly. These practices transform regulatory complexity from an obstacle into a competitive advantage.
For companies developing AI tools in diagnostic imaging, clinical decision support, or other high-impact healthcare applications, the US market represents enormous potential.Navigating the path to that market requires expertise, planning, and persistence—but the destination rewards the journey.
FAQs
Our frequently questions
Because the US offers faster validation and commercialisation pathways for AI-powered medical devices, despite its regulatory complexity, whereas EU frameworks (MDR, GDPR) often slow down market entry and scale.
AI medical devices are regulated through a combination of FDA oversight (SaMD and clinical validation), OHRP requirements for human subject research, and IRB approvals at the institutional level, creating a layered regulatory system distinct from EU ethics committees.
Although AI developers are usually not HIPAA-covered entities, access to patient data depends on HIPAA-compliant disclosures by hospitals and clinics, requiring proper authorisations, IRB waivers, or justified de-identification strategies.
Only if secondary use is explicitly planned and authorised from the outset; data collected for research or treatment cannot automatically be reused for model training without appropriate consent, IRB approval, or legally compliant de-identification.

Find out how iliomad can help your company.
Only visible in production

Data Protection in Clinical Trials: Governing AI-Assisted Data Flows Under GDPR
AI intensifies clinical trial data protection, requiring end-to-end governance and inspection-ready controls.
Weekly Digest: EU AI Act Prohibitions, GDPR Enforcement and Clinical Trial Developments - Week of 22 June 2026
AI healthcare rules tighten as EU, US and GDPR developments accelerate.
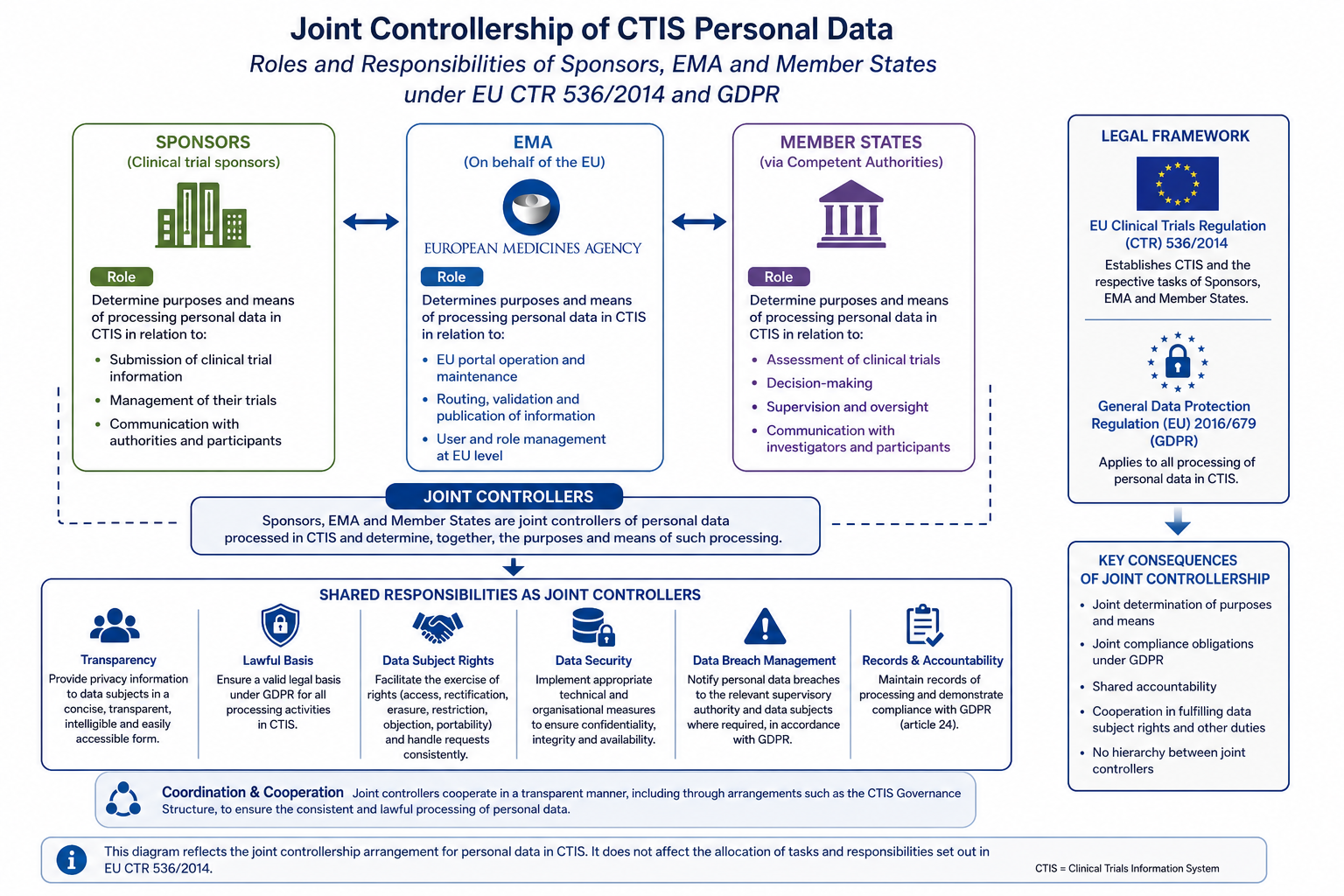
CTIS Data Protection: Joint Controllership, GDPR Obligations and Sponsor Responsibilities
Understand CTIS data protection obligations under EU CTR 536/2014 and GDPR. Learn how the Joint Controllership Arrangement allocates roles among sponsors, EMA and Member States.
