



Summary
Data protection in clinical trials has become substantially more complex as artificial intelligence enters every stage of the trial lifecycle, from site monitoring and safety signal detection to medical writing and inspection preparation. Sponsors operating under the General Data Protection Regulation (Regulation (EU) 2016/679, GDPR) must now govern not only source databases but every pathway through which AI tools process, transform or summarise personal data. This article sets out a practical framework for end-to-end data flow integrity, covering legal bases, cross-border transfers, CRO accountability, country-specific requirements and the operational steps needed to keep global studies inspection-ready.
Why Data Flow Integrity Matters for Data Protection in Clinical Trials
Data protection in clinical trials is not confined to securing a single database. It covers every point at which personal data is captured, transmitted, transformed or stored across the trial lifecycle. When AI tools are introduced, each transformation step creates a new compliance obligation, because Art. 5(1)(f) GDPR requires personal data to be processed in a manner that ensures appropriate security, including protection against unauthorised processing.
Traditionally, data integrity governance focused on the accuracy of individual data points inside an Electronic Data Capture (EDC) system. AI changes that scope fundamentally. An AI model used for monitoring anomaly detection may ingest raw site data, produce a derived risk score and feed that score into a Clinical Trial Management System (CTMS) or an electronic Trial Master File (eTMF). At each node of that pathway, personal data is being processed within the meaning of Art. 4(2) GDPR, and the controller (typically the sponsor) remains accountable for the entire chain. This operational reality sits at the heart of our data protection services for clinical trials.
ICH E6(R3), the Good Clinical Practice guideline revised in 2023, reinforces this view by requiring sponsors to maintain oversight of all computerised systems used in a trial, including those operated by CROs and vendors. The 2024 FDA guidance on electronic systems in clinical investigations reaches a parallel conclusion: sponsors must be able to reconstruct how trial decisions were made, including decisions influenced by AI-generated summaries or assessments.
From a GDPR perspective, the inability to reconstruct an AI-assisted data pathway is not merely a scientific concern. It is a potential breach of the accountability principle (Art. 5(2) GDPR) and may undermine the legal validity of the processing if the sponsor cannot demonstrate that appropriate safeguards were in place.
What Legal Basis Applies When AI Processes Trial Participant Data?
The legal basis for processing clinical trial data under GDPR depends on the purpose of the processing, and AI use does not create a new or separate legal basis. For interventional trials, the two most commonly applicable bases are Art. 6(1)(a) (explicit consent) combined with Art. 9(2)(a) for special category health data, and Art. 9(2)(j) (scientific research), which must be read alongside Art. 89 GDPR and applicable national law.
Health data collected from trial participants constitutes special category data under Art. 9(1) GDPR, which means processing is prohibited unless one of the conditions in Art. 9(2) is satisfied. When AI tools process that data, whether to detect safety signals, generate protocol deviation summaries or assist medical writers, the same legal basis applies. There is no general exemption for AI-assisted processing.
The research derogation under Art. 89 GDPR allows member states to provide limited derogations from certain data subject rights (Art. 15, 16, 18 and 21) when data are processed for scientific research purposes, provided appropriate safeguards such as pseudonymisation are applied. Art. 89 does not exempt sponsors from the obligation to conduct a DPIA under Art. 35 GDPR, which is triggered whenever processing is likely to result in a high risk to individuals, a threshold that large-scale health data processing in AI-assisted environments almost always meets.
The EU Clinical Trials Regulation 536/2014 (EU CTR), fully applicable since January 2023, further requires the clinical trial protocol to include a description of the measures taken to protect personal data. Protocol Section 13 is the standard location for this description, and its content must be consistent with the GDPR legal basis identified in the sponsor's Record of Processing Activities.
How Sponsors Should Govern CRO and Vendor Data Flows Under GDPR
Sponsors must treat every CRO or technology vendor that processes personal data on their behalf as a data processor under Art. 28 GDPR, which requires a written contract specifying the subject matter, duration, nature and purpose of the processing, the type of personal data and the obligations of both parties. This is a mandatory legal requirement, not a best practice. Recent enforcement, analysed in our review of vendor GDPR obligations in clinical trials, shows how quickly undisclosed sub-processing becomes a sponsor liability.
Establishing clear data ownership and processor accountability
The sponsor acts as the data controller and retains full accountability for the lawfulness of all processing, including processing carried out by CROs. Where a CRO uses sub-processors, including AI platform providers, the DPA must either grant general written authorisation for sub-processing subject to the same data protection obligations (Art. 28(4) GDPR) or require specific prior written consent for each sub-processor.
In practice, governance of AI-assisted workflows requires four things:
• Documented provenance: the sponsor must be able to identify the origin of every data element, including AI-generated summaries, and trace any transformation applied to it.
• Version control: AI model versions used to produce regulatory-relevant outputs must be recorded, because a change in model version is a change in processing that may require a DPIA update.
• Human review records: where AI outputs inform safety narratives, deviation assessments or inspection responses, the record must show that a qualified person reviewed and accepted or overrode the AI output. This aligns with the ALCOA+ requirement for attributability.
• Access controls: Art. 32 GDPR requires technical measures commensurate with the risk. For AI platforms processing health data, this includes role-based access, encryption at rest and in transit, and audit logs that cannot be altered retroactively.
Risk-based validation of AI systems
ICH E6(R3) requires computerised systems used in clinical trials to be validated for their intended purpose. When that purpose includes processing personal data, validation records become part of both the quality system and the data protection governance documentation. A DPIA conducted under Art. 35 GDPR should reference system validation status and identify residual risks that require additional safeguards. Our AI compliance services help sponsors map these controls across in-house and vendor systems.
Cross-Border Transfer Obligations When AI Tools Are Hosted Outside the EEA
Many AI platforms used in clinical research are hosted by providers based in the United States or other third countries. Transferring personal data to a third country is permissible under GDPR Chapter V only if an adequate level of protection is ensured. Sponsors must identify the applicable transfer mechanism before any data leaves the European Economic Area (EEA). Three primary mechanisms are available:
• Adequacy decisions: issued by the European Commission under Art. 45 GDPR, covering countries such as the United Kingdom (adequacy decision adopted June 2021, currently under review), Japan and, since July 2023 for participating organisations, the United States under the EU-US Data Privacy Framework.
• Standard Contractual Clauses (SCCs): adopted by Commission Decision 2021/914/EU, providing a contractual safeguard when no adequacy decision covers the recipient.
• Binding Corporate Rules (BCRs): under Art. 47 GDPR, applicable within corporate groups.
For clinical trials, the most frequently used mechanism remains the SCCs, because trial data often flows to multiple vendors in multiple countries for which no adequacy decision exists. A Transfer Impact Assessment (TIA) must accompany each set of SCCs to evaluate whether the law and practice of the destination country may impair the effectiveness of the safeguards, as required by the European Data Protection Board (EDPB) Recommendations 01/2020.
AI tools that process trial data in the cloud add a further complexity: data may be processed in jurisdictions different from those where the cloud servers are nominally located, depending on disaster recovery and load-balancing configurations. Sponsors must require vendors to disclose all sub-processing locations and ensure that the transfer mechanism covers each one.
Country-Specific Focus: Regulatory Formalities Across Key Jurisdictions
France: CNIL and MR-001
France requires interventional clinical trials to comply with the Méthodologie de Référence MR-001, a reference methodology published by the Commission Nationale de l'Informatique et des Libertés (CNIL) that sets out the conditions under which sponsors may process health data without a specific CNIL authorisation, provided they commit to MR-001 in full. Where AI tools introduce processing operations that fall outside the scope of MR-001, such as secondary analysis of aggregated participant data for model training, a specific CNIL authorisation under Art. 66 of the French Data Protection Act (Loi Informatique et Libertés) may be required. The CNIL published updated guidance on AI in health research in 2024, emphasising that automated processing of health data remains subject to the prohibition in Art. 9 GDPR and that pseudonymisation alone does not remove that obligation. See our guide to the MR-001 reference methodology for the operational detail.
Germany: state data protection laws and DPO obligations
Germany implements the research derogation through the Landesdatenschutzgesetze (state data protection laws), because health research regulation is partly a matter of Länder competence. Sponsors opening sites in Germany must verify the applicable state law for each site. All organisations processing health data on a large scale in Germany must appoint a Data Protection Officer (DPO) under Section 38 of the Bundesdatenschutzgesetz (BDSG), which lowers the threshold compared with Art. 37 GDPR. The DPO must have expert knowledge of data protection law and practice: a clinical operations professional without dedicated data protection qualifications does not satisfy this requirement, which is why many sponsors appoint an outsourced DPO with sector expertise.
United Kingdom: UK GDPR and ICO guidance
Following the UK's departure from the European Union, the UK GDPR (retained in domestic law by the Data Protection Act 2018) mirrors the EU GDPR in most substantive respects. The Information Commissioner's Office (ICO) issued guidance in 2023 on AI and data protection, requiring organisations carrying out AI-assisted processing to conduct an Art. 35-equivalent Data Protection Impact Assessment and to document the lawful basis for any solely automated decision-making. For clinical trials, the UK's existing adequacy decision, issued by the EU Commission in June 2021, facilitates data transfers from EU trial sites to UK-based sponsors, but sponsors must monitor the scheduled review of that decision.
Spain: AEPD and biomedical research
Spain's Agencia Española de Protección de Datos (AEPD) published a code of conduct for biomedical research in 2022 that addresses the use of AI in health data processing. Spain's Organic Law 3/2018 on Personal Data Protection requires explicit consent for the secondary use of health data for research purposes, unless the research falls within a recognised public interest framework. Sponsors using AI tools that aggregate data from Spanish sites for model improvement or population analytics must assess whether that secondary processing is compatible with the original research purpose under Art. 5(1)(b) GDPR.
EMA and EU CTR 536/2014
The EU CTR (Regulation (EU) No 536/2014) requires sponsors to include a data protection section in the clinical trial protocol, and the EMA's assessment of the trial application includes a review of that section. The EMA's clinical data publication policy (Policy 0070) further requires that clinical study reports submitted for marketing authorisation be made available for public access in a form that protects personal data. Sponsors using AI to generate or summarise clinical study reports must ensure that AI-assisted outputs can be appropriately redacted before publication and that the redaction process itself does not introduce privacy risks through residual metadata.
Comparison of the Key Instruments Governing Data Protection in AI-Assisted Trials
The regulatory framework governing data protection in AI assisted clinical trials is not based on a single instrument, but on a combination of overlapping legal, regulatory and good clinical practice requirements. A single AI assisted study may simultaneously trigger obligations under the GDPR, including legal basis, DPIA, accountability and data subject rights requirements; the EU Clinical Trials Regulation, including protocol level data protection disclosures and public transparency rules; ICH E6(R3) GCP, which reinforces sponsor oversight and validation of computerised systems, including AI tools; and the EU SCCs, where trial data is transferred to third countries. Country specific rules may also apply, such as France’s MR 001 methodology for interventional research or Germany’s BDSG rules on DPO appointment. In parallel, the EU AI Act introduces risk classification and high risk obligations for certain medical AI systems, while ALCOA+ principles remain essential to ensure that all trial data, including AI generated outputs, is attributable, legible, contemporaneous, original, accurate and traceable.
FAQs
Our frequently questions
GDPR does not apply to data that is truly anonymous, meaning data that cannot be re-identified by any means reasonably likely to be used (Recital 26 GDPR). However, AI outputs derived from personal data are rarely fully anonymous because they may retain statistical properties that allow indirect identification, particularly in small patient populations. Sponsors should conduct a re-identification risk assessment before treating any AI-generated output as anonymous.
A DPIA is mandatory under GDPR Article 35 whenever processing is likely to result in a high risk to the rights and freedoms of individuals. Large-scale processing of health data, which is a special category under Article 9 GDPR, is explicitly listed as a trigger category in Article 35(3)(b). In practice, any clinical trial using AI to process participant health data at scale will meet this threshold, so a DPIA is required before processing begins.
Undisclosed sub-processing by a CRO is a breach of the Data Processing Agreement required by Art. 28 GDPR and may breach the EU CTR 536/2014 if it affects the integrity of trial records. The sponsor, as data controller, remains liable to supervisory authorities for the CRO's non-compliant processing. Sponsors should include contractual provisions requiring prior written notification of any new AI tool that processes personal data, with a right to object before deployment.
Legitimate interests cannot override the special category prohibition in Art. 9(1) GDPR for health data. Sponsors must identify a specific condition in Art. 9(2), most commonly Art. 9(2)(j) for scientific research or Art. 9(2)(i) for public health, alongside an Art. 6 basis. Legitimate interests under Art. 6(1)(f) may be relevant for ordinary personal data (such as contact details of site staff) but not for health data.
Art. 5(2) GDPR requires the controller to demonstrate compliance with all processing principles. For AI tools, this means recording the model version in use at each relevant time point, the training data used, the validation status under ICH E6(R3) and any change that could affect the nature of the processing. This documentation should sit within the Record of Processing Activities under Art. 30 GDPR and be cross-referenced in the eTMF.
The EU AI Act (Regulation (EU) 2024/1689) applies in addition to GDPR, not instead of it. AI systems used in clinical research can fall into the high-risk category where they act as, or as a safety component of, a medical device, or where they materially influence clinical decisions. High-risk systems carry obligations on risk management, data governance, logging, human oversight and post-market monitoring. Sponsors should classify each AI tool early, because the obligations and timelines differ from GDPR and several provisions phase in through 2026, as we set out in our briefing on the EU AI Act for healthcare.

Find out how iliomad can help your company.
Only visible in production
Weekly Digest: EU AI Act Prohibitions, GDPR Enforcement and Clinical Trial Developments - Week of 22 June 2026
AI healthcare rules tighten as EU, US and GDPR developments accelerate.
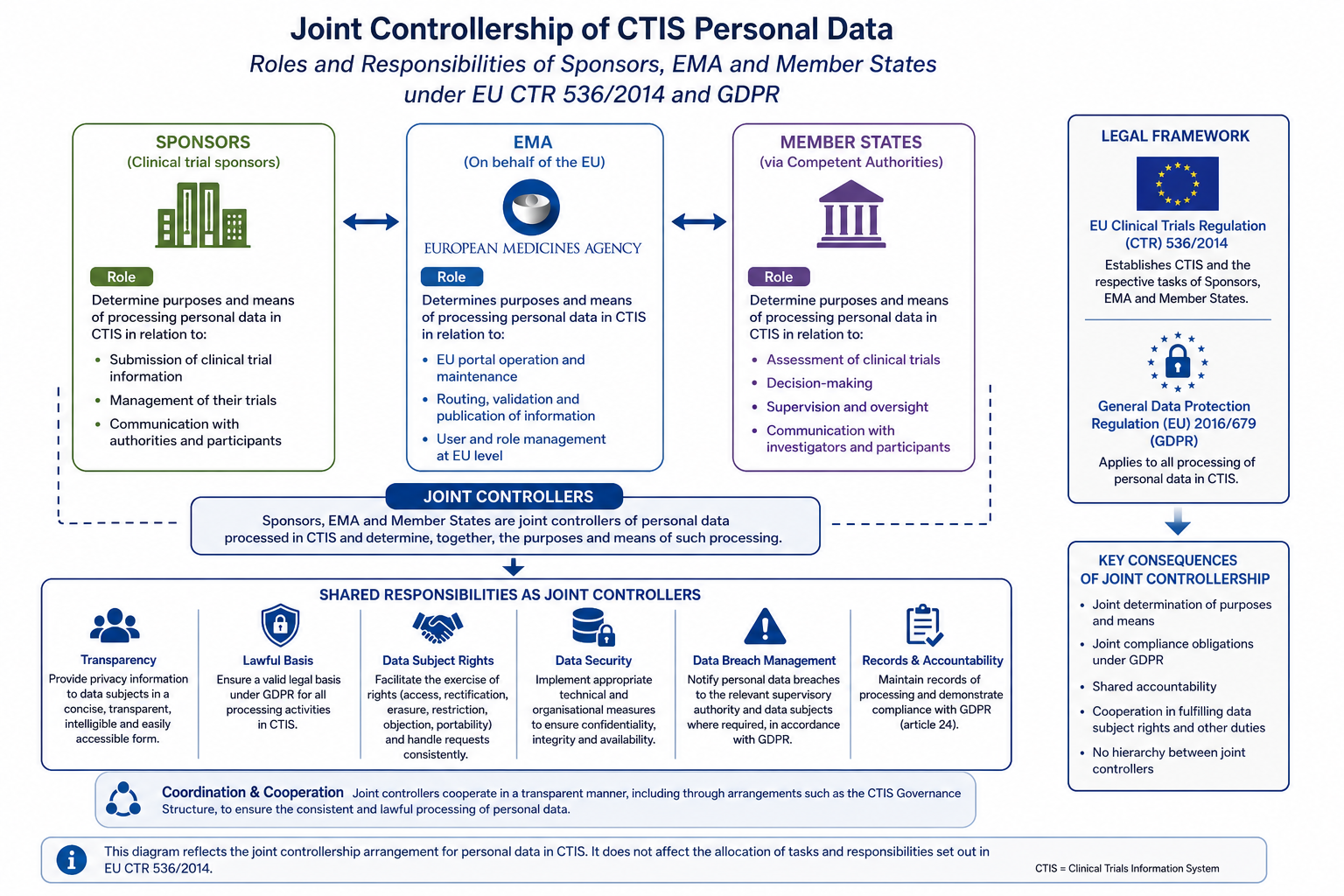
CTIS Data Protection: Joint Controllership, GDPR Obligations and Sponsor Responsibilities
Understand CTIS data protection obligations under EU CTR 536/2014 and GDPR. Learn how the Joint Controllership Arrangement allocates roles among sponsors, EMA and Member States.

Pseudonymisation Clinical Trials: CNIL's Enforcement Clarifications and What They Mean for Life Sciences Organisations
CNIL's 2026 enforcement action clarifies the line between pseudonymisation and anonymisation in clinical trials. Learn the GDPR implications and recommended practices.
